

中国粉体网讯 自2015年以来,国内新药政策环境发生重大变化,多项支持性政策相继出台,国内新药审评审批制度逐渐在向法规体系成熟的欧美国家靠近。新药加速上市政策的出台促使审评审批流程大大简化。同时,借鉴美国成熟的加速上市通道,国家药品监督管理局制定了国内加速上市程序,包括突破性疗法、附条件上市、优先审评。下文将对加快新药上市的重要政策进行梳理和介绍。
新药加速上市政策变革
1取消三报三批,允许国内外同步开发
2017年,国家药监局颁布《调整进口药品注册管理有关事项的决定》(以下简称决定),取消了针对进口药在国内注册的多项限制。其中,取消三报三批对进口药注册、中国新药与国际接轨的重要性不言而喻。
1. 三报三批是什么?
“三报三批”是指:进口产品以国际多中心(IMCT)申报上市需完成三个步骤:
①报IMCT,获得IMCT批件
②报CTA,进口药注册临床试验批件
③报NDA、批进口注册
2.三报三批对新药进口注册有哪些影响?
“三报三批”要求企业在已取得满足中国注册要求的中国患者数据的前提下,重复按临床申报流程申请批准临床批件,临床批件获得后再按上市申报流程申报上市。企业在步骤②中重新递交已被审评过的资料,相当于多走一轮2年或更久的临床试验审批程序,由于当时药品审评审批积压严重,重新排一次队直接导致产品整体上市时间平均延长3-5年。
3.与之前的做法相比,《决定》调整的事项主要有三个方面:
允许同步研发申报: 当时实施《药品注册管理办法》(以下简称《注册办法》)要求,境外申请人向总局申请开展MRCT的药物,应当是已在境外注册或者已经进入II期或III期临床试验。《决定》实施后,除预防用生物制品外,允许在中国境内外同步开展Ι期临床试验。
优化注册申报程序: 《注册办法》中MRCT申报及审评审批是相对独立的程序,开展MRCT的药品申请进口的,需要按照进口药品注册程序申报。《决定》实施后,开展MRCT的药品申请进口,符合《药品注册管理办法》及相关文件要求的,可以直接提出进口上市注册申请。
取消部分进口药品在境外上市的要求: 具体而言,对于提出进口临床申请、进口上市申请的化学药品新药以及治疗用生物制品创新药,取消应当获得境外制药厂商所在生产国家或者地区的上市许可的要求。
2药品加快上市注册程序,让新药开发降本提速
新药开发策略核心关注点是加快临床开发、加快审评审批。2005年11月,国家药监局发布《国家食品药品监督管理局药品特别审批程序》(局令第21号),就特别审批程序的适用范围以及工作程序进行阐述。
2020年7月,国家药监局发布《突破性治疗药物审评工作程序(试行)》等三个文件的公告(2020年第82号)。2020年11月,CDE发布《药品附条件批准上市技术指导原则(试行)》。结合以上文件,国内“突破性治疗药物程序”、“附条件批准程序”、“优先审评审批程序”“特别审批程序”的适用范围及申请阶段总结如下:
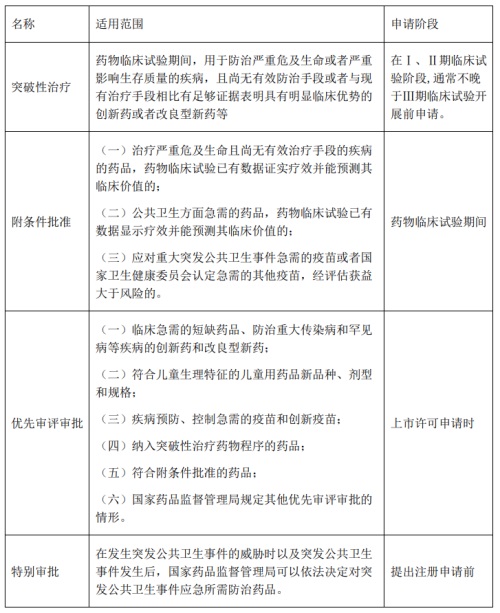
以上四个程序同时适用于国内新药、进口新药,在加速新药开发上市的过程中,适用的情况存在一定差异。由于特别审批程序仅适用于应对公共卫生事件的药品,实践中应用较少。这里仅介绍其他三种常用程序:
“突破性治疗药物程序”:具有加速临床开发进度、上市审评审批速度的双重作用。通常使用早期临床数据进行该资格申请。对于获得突破性治疗资格的新药,CDE将优先处理相关沟通交流,加强临床试验开发的指导,确保开发策略正确。另外,在申报上市环节,该类药品可同时适用优先审评程序,从而缩短审评时间。需要强调的是,一种药物获得突破性治疗资质并不意味着可以依据不完整的临床研究资料批准该产品上市。监管机构或者有经验的审评人员的早期加入,是为了帮助申请人及时发现研究中存在的问题,提出巧妙灵活又能满足监管要求的研究策略,以尽可能少的样本量和研发时间获得足以证实临床价值的安全有效性数据,减少因关键性研究方案设计缺陷导致的失败。突破性治疗药物的身份可以让产品在临床研究期间得到更多的关注,吸引到更多的社会资源和支持。集中力量服务于具有最迫切临床需求的患者人群,将有限的审评和临床资源向最具潜力的优势产品倾斜,是以患者利益为中心和提高社会公平性的具体体现。
“附条件批准程序”: 在加速临床开发进度以及上市方面,优势最为明显。但要求该疾病存在可以或可能预测获益的替代终点、中间终点。可以基于II期或III期临床试验替代终点或III期试验中间终点的数据申请附条件上市,上市后继续开展或完成III期临床试验以确定药物的安全性和有效性。和常规上市的药物相比,总体上市时间至少提前2至3年。国内附条件批准药物中,绝大多数为抗癌新药,关键性临床的设计主要以单臂II期研究为主。采用III期试验期中数据(ORR为主要终点)设计的较少,该设计通常采用ORR\OS双终点设计,由于ORR随访时间较短,通常为几个月,在第一次期中分析时,如已经达到主要终点。即可基于ORR终点数据申请附条件批准,在上市后完成OS的随访即可。
“优先审评审批程序”:该程序仅加速审评过程,不能加速药品临床开发进度。对纳入优先审评审批程序的药品上市许可申请,审评时限为130日,其中临床急需的境外已上市境内未上市的罕见病药品审评时限为70日。
小结
目前,国内已有多家药企开发的新药享受到这些政策红利,包括豪森的奥美替尼、百济神州的泽布替尼等,均是利用单臂关键二期申请附条件批准,加快上市许可的同时节省了新药开发成本。如何利用这些政策红利已成为新药能否快速上市的关键因素,上市时间也决定一个新药在同类药物中的市场地位。
参考文献:
1.2017《关于调整进口药品注册管理有关事项的决定》
2.2005《国家食品药品监督管理局药品特别审批程序》(局令第21号)
3.2020《突破性治疗药物审评工作程序(试行)》等三个文件的公告(2020年第82号)
4.2020 《药品附条件批准上市技术指导原则(试行)》
5.邹丽敏,杜 瑜,齐玥丽, 唐凌,杨志敏,抗肿瘤新药突破性治疗药物认定的标准及考虑,中国临床药理学杂志,第 37 卷 第 11 期
(中国粉体网编辑整理/青黎)
注:图片非商业用途,存在侵权告知删除














