中国粉体网讯
研究背景
近年来,高浓缩电解液(HCE)、局部高浓缩电解质(LHCE)、和弱溶剂化电解液(WSE)的新设计概念将锂金属负极的循环可逆性带入了一个新时代,其中的核心谜团是阴离子衍生的SEI。这种独特的SEI化学具有更有利的特性,包括空间均匀性、化学惰性和机械鲁棒性,以减轻SEI本身的损伤和生长。此外,在阴离子衍生的SEI下,Li沉积方式可以从针状形态定制成更均匀的颗粒形状大大减少了电隔离的死锂的数量。这两个方面的共同贡献有助于大大提高工作的锂金属负极的循环可逆性。
虽然这些电解质设计的新进展明确地提高了锂金属负极的室温性能提高到一个全新的水平,但工作电池实际上受到不同温度条件的影响。深入了解温度依赖的Li沉积/溶解行为,以及揭示温度依赖的库仑效率的关键决定因素,具有重要意义。在这种背景下,最近的一些研究已经表明,提高工作温度在一个合理的范围内可以促进锂金属负极的性能,通过界面和动力学的好处,与之形成鲜明对比的是,锂金属负极的零下温度性能远不令人满意,其主要瓶颈在于工作温度降低时极化程度较大,可逆性较差(图1)。实际上,锂沉积的完整图像包括体离子输运、界面锂离子(Li+)脱溶、Li+在SEI中的扩散和电子转移。如Vogel–Tammann–Fulcher(VTF)方程和阿伦尼乌斯方程所述,这四种过程的动力学速率本质上都依赖于温度,而最电阻步骤(速率决定步骤)仍存在争议。此外,工作温度的降低通常伴随着锂沉积/溶解的可逆性显著降低,这也是一些表现出优越的室温性能的电解质的情况。揭示低温锂沉积过程中的动力速率决定因素,阐明锂金属负极的极化问题和可逆性问题之间的相互作用,是构建稳定低温锂金属电池的首要步骤。
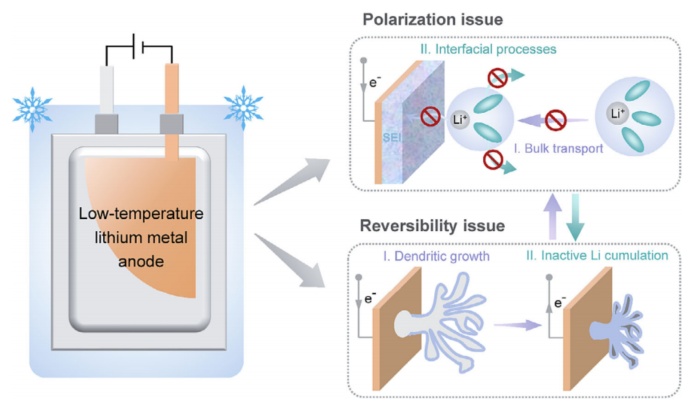
图1稳定低温金属锂负极的主要瓶颈,即极化问题、可逆性问题以及这两个因素如何相互作用的示意图。
成果简介
近日,北京理工大学黄佳琦教授对锂沉积过程中的过电位属性进行了分解,并建立了动力学过电位支配、SEI化学的动态演化和相应的Li可逆性之间的相互作用。通过采用典型的LHCE作为模型系统,揭示了离子浓度梯度发挥压倒性的作用在极化正极锂沉积过程在零下温度下,这不仅直接限制工作电池的功率输出,而且严重降低锂沉积均匀性的有限的阴离子分解动态SEI形成。受研究结果的启发,将低温电解质的设计合理解耦,同时抑制工作过电位,为SEI的动态形成提供可持续的协调阴离子供应。因此,在1.0mAcm-2下,-20°C的Li沉积/溶解CE明显从95.93%提高到98.40%,动力学过电位降低,在实际的Li|LiNi0.5Co0.2Mn0.3O2(NCM523)电池中进一步证明寿命延长了3倍以上。该工作以“Unlocking the Polarization and Reversibility Limitations for StableLow-Temperature Lithium MetalAnodes”为题发表在small structures上。
研究亮点
(1)探究了在不同温度不通电流密度下的局部高浓电解液体系里的锂沉积行为。
(2)将锂金属沉积过电位分解,通过时间分辨的弛豫测量分析出浓度过电位、界面反应过电位、欧姆过电位,并探究收温度、电流密度对极化电位的影响。
(3)通过核磁定量的探究在不用的电流密度下的电解质中DME与FSI-的消耗,在较大的Li+浓度梯度下,Li+-阴离子相互作用减弱,不利于阴离子衍生的SEI形成。
(4)合理的设计出一款低过电位和高锂可逆性的低温电解质的设计策略,引入了微量(0.05M)的强配位硝酸锂(LiNO3),以补偿Li+-阴离子的聚集损失。
图文导读
局部高浓电解液中的低温锂沉积行为
1.0MLiFSI溶解于DME/TTE电解质标记为基线LHCE,此电解液被广泛报道但低温性能仍有待研究。首先,系统地比较了Li|Cu电池在25和-20°C时的Li沉积行为。如图2a、b所示,平均CEs小电流密度0.2mAcm-2时比较类似,而当电流密度提升到1.0mAcm-2平均CE的差异变大。在25°C时Li+可逆沉积/溶解的库仑效率为99.12%,-20°C的库仑效率降低至95.93%,这不利于长寿命可充电电池。图2c、d进一步记录了不同温度和电流密度下的Li沉积形态,以找出不同库仑效率的原因。在0.2mAcm-2的小电流密度下,Li沉积物在25和-20°C下均呈现出良好的大颗粒状形态(图2c),这与它们在Li|Cu电池中获得的库仑效率很一致(图2a)。当电流密度增加到1.0mAcm-2,即使在25°C下可以保持颗粒状锂沉积形态,当工作温度降低到-20°C时,它变成了具有高纵横比针状形态的高度树突状结构(图2d),Li沉积行为的急剧转变导致可逆性明显下降。
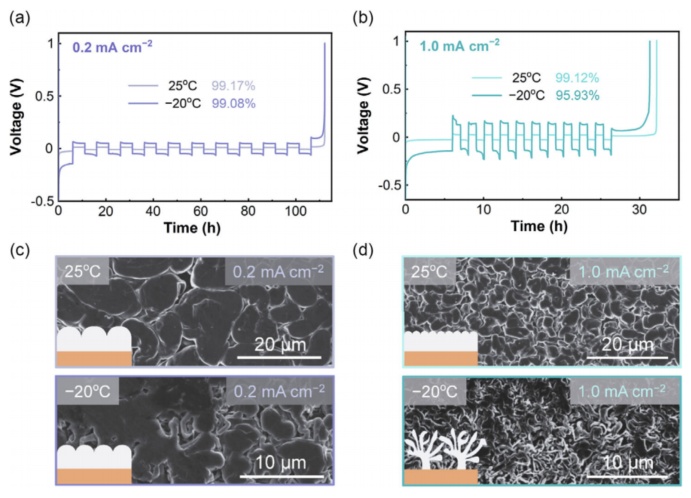
图2LHCE电解液中不同温度和电流密度下的锂沉积行为,包括不同温度下Li|Cu半电池在(a)0.2mAcm-2和(b)1.0mAcm-2电流密度下的平均库仑效率,和不同温度下Li|Cu半电池在(c)0.2mAcm-2和(d)1.0mAcm-2电流密度下的的沉积形貌。在(a,b)循环和(c,d)沉积中的总容量为1.0mAhcm-2。
在研究界广泛承认的是,Li的沉积行为主要由SEI控制,而SEI的形成过程直接由Li+的溶剂化结构决定。基于这一观点,通过分子动力学(MD)模拟,直观地研究了与温度相关的Li+溶剂化结构。模拟结果如图3a、b,降低温度提高了阴离子-阳离子交互强度,这可以通过在Li+的初级溶剂化壳中Li+和FSI-之间的Li-O相互作用更强的峰验证,因此将温度从25降低到-20°C后,FSI-的平均配位数增强(从2.61到2.76)。此外,特定Li+配位态的微观统计显示,DME分子单独溶剂化的Li+百分比(3-0-0)从5.0%显著下降到0.8%(图3c,d)。根据已建立的理论,一个更强的阴离子-阳离子配位将促进更多的阴离子分解,以促进阴离子衍生的SEI的形成。为了证实这一点,我们进行了电化学阻抗谱(EIS)表征,以探测在不同温度下长期静置以形成静态SEI的对称Li电池的界面电阻,-20°C时的电池电阻低于25°C时的电阻,而由于SEI富无机性质,两种SEI具有相似的离子激活能(Ea)。以上观察明显强调,静态Li+溶剂化结构的变化不应该是决定锂沉积方式转变的主要原因,而是更可能是由于实际工作的Li负极的动态相关界面行为导致的。
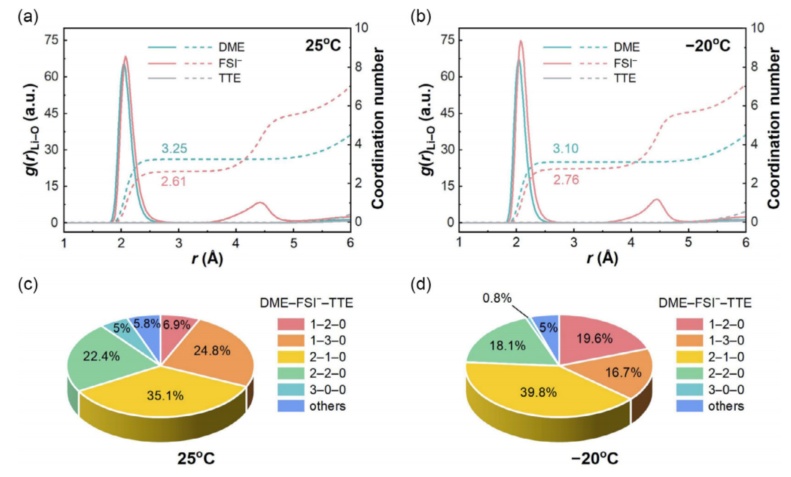
图3通过MD模拟了具有温度依赖性的Li+溶剂化结构。根据(a)25°C和(b)-20°C,通过MD模拟轨迹计算的Li-ODME,Li-OFSI-和Li-OTTE对的径向分布函数(实线)和配位数图(虚线),(c,d)是不同Li+配位结构的相应统计。
低温下锂沉积的动态研究
由上述演绎,一个时间分辨瞬态弛豫测量来分析动态过电位属性在低温锂金属沉积(图4),其中一个简短的弛豫步骤(10.0s)是间歇地引入探头连续恒流锂沉积过程中区分浓度过电位(ηconc)和iR下降(图4a、b)。重要的是,iR下降被定义为弛豫步骤前的最终电位值与指定静置时间后的弛豫电位值之间的突然电位下降。合理选择界面电荷转移过程的弛豫时间为时间常数(τ)的5倍,以确保界面反应过电位(ηreac)完全释放,使其合并到iR下降中。然后,考虑到ηohm通常不受锂沉积深度的影响,总iR可以被分解为欧姆过电位(ηohm)和ηreac。在图4c、d中可以清晰的分解。结果似乎与之前的认识相反,即界面过程(Li+通过SEI或Li+脱溶态)是低温负极的动力学速率决定步骤;这里发现,在-20°C的Li沉积中,在0.2和1.0mAcm-2下,ηconc对总过电位的贡献最大(图4c、d)。值得注意的是,当将Li沉积温度升高到25°C时,过电位的支配地位转移到了ηreac,这可以从温度从-20°C(0.21mScm-1)提高到25°C(0.70mScm-1)时离子电导率的显著变化中解释。此外,我们发现,无论电流密度(图4c,d)和温度(如何,ηreac都随着Li沉积容量的增加而降低。其主要原因是随着锂沉积深度的增加,活性表面积逐渐增大,从而降低了界面上电化学反应的电阻和过电位。进一步进行了COMSOL模拟,从理论上考察了低温锂沉积条件下离子浓度的演化(图4e、f)。当电流密度从0.2mAcm-2,增加到1.0mAcm-2时,Li+载流子的消耗和补充明显失衡,导致连续沉积锂后负极表面的离子浓度降低了近一半(图4f)。
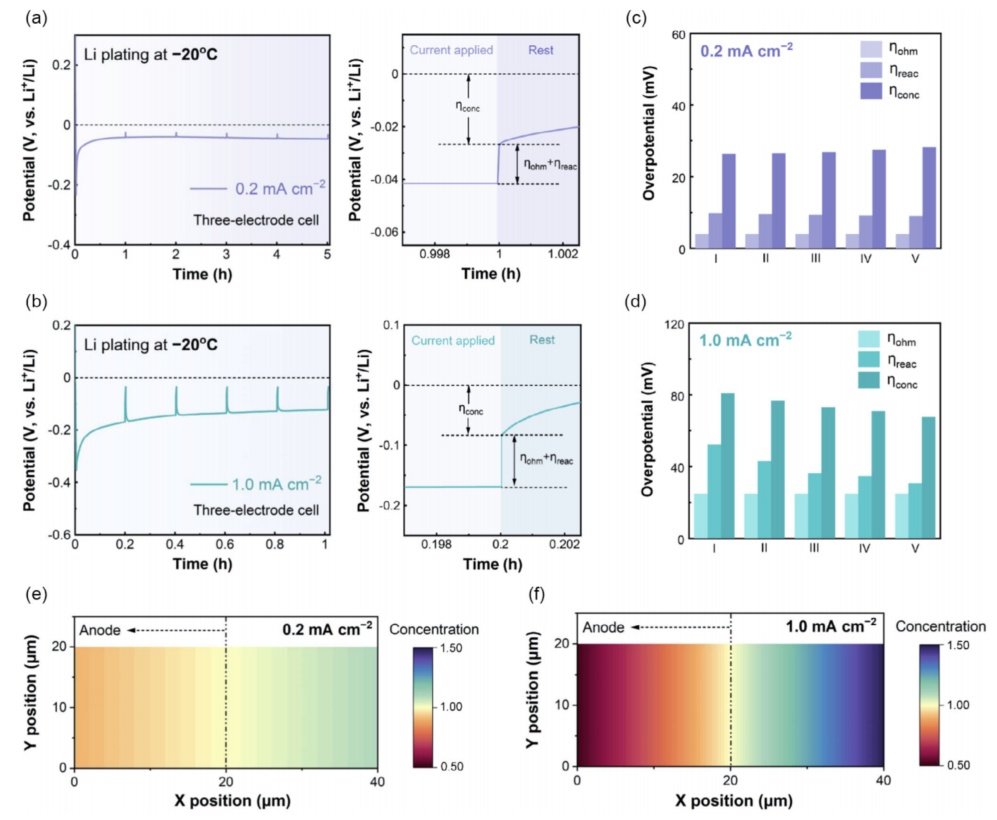
图4在(a)0.2mAcm-2和(b)1.0mAcm-2下,三电极时间分辨瞬态弛豫测量对低温锂沉积的过电位属性进行分解在(c)和(d)中得出了不同的的ηohm、ηCT和ηconc在(e)0.2mAcm-2和(f)1.0mAcm-2下沉积1.0mAhcm-2后,电解液中离子浓度梯度的COMSOL模拟结果。
一方面,如Sandy的时间模型所述,Li+浓度的耗尽有可能引发Li树突的生长。另一方面,局部Li+浓度的演化应该会强烈地改变原来的Li+阴离子相互作用,而在Li+浓度动态降低的情况下,Li+阴离子相互作用逐渐减弱,预计将极大地影响SEI的动态形成过程。为了证实后一点,我们通过核磁共振(NMR)技术定量地研究了阴离子和溶剂对SEI渐进形成的动态消耗。本研究有意利用无SEI、结构稳定的Li7Ti5O12电极为Cu上的锂沉积提供Li源,以必然排除对电极的电解质消耗(图5a)。分别在19F和1HNMR下(图5b)测定了Cu|Li7Ti5O12电池10个循环后的电解质。通过追踪FSI-(19F谱)和DME(1H谱)相对于内参试剂的峰值强度演化,可以很容易地定量盐和溶剂的动态消耗。如图5c所计算,在0.2mAcm-2的电流密度下的FSI-消耗(9.5%)比DME(7.2%)更显著,而在1.0mAcm-2下选择性消耗DME,因为DME的损失(11.4%)显著高于FSI-(3.8%)。通过x射线光电子光谱(XPS)进一步检测了在0.2和1.0mAcm-2下Li沉积后的SEI化学性质。如图5d、e所示,SEI形成于在0.2mAcm-2富含FSI-的分解产物LiF、Li2O,Li3NandLi2S。然而,当电流密度增加到1.0mAcm-2时,这些阴离子衍生的SEI物种的数量明显减少上述结果生动地表明,动态电解质消耗SEI形成反应和由此产生的SEI可以调控的实时Li+-阴离子交互界面,在较大的Li+浓度梯度下Li+-阴离子结合强度减弱,不利于阴离子衍生的SEI的形成。
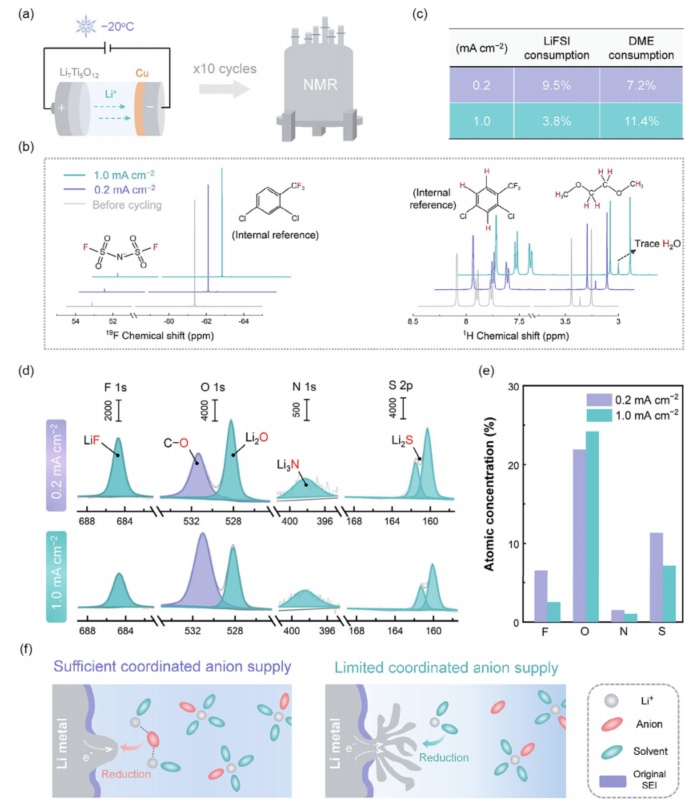
图5通过核磁共振技术测定阴离子和溶剂的消耗,包括(a)表征程序的示意图,(b)未循环和循环电解质的19F和1HNMR谱,以及(c)计算出的FSI-和DME的消耗比。在XPS下对-20°C的Li沉积物进行SEI化学分析,包括(d)F1s、O1s、N1s和S2p光谱,以及(e)相应的原子浓度。这些数据是在120s的溅射后获得的。(f)在协调阴离子供应充足和有限的情况下,动态SEI修复过程的示意图,这分别导致颗粒状和树突状Li沉积。
为了进一步证实Sandy的时间模型或动态界面化学模型是决定Li沉积和电池可逆性的决定因素,我们设计了一个通过调整DME的体积比例到TTE的对照实验。当提高体积/TTE从1/2到2/1,离子电导率-20°C可以提高38.1%到0.29mScm-1。受益于增强盐解离,有利于明显的缓解极化锂沉积/剥离。然而,这种加速Li+扩散的好处并没有导致对Li可逆性的任何预期的增强。相反,平均库仑效率甚至从接近96%下降到95.31%,这可以归因于一旦降低了体积电解质中有效的Li+浓度(DME中的Li+浓度),Li+-阴离子相互作用不足,导致阴离子还原。
在这里,我们可以安全地得出结论,实时Li+浓度依赖的Li+-阴离子相互作用和相应的动态SEI形成途径是决定Li沉积行为,从而决定电池低温下沉积行为可逆性的关键因素。当配位阴离子供应充足时,裂解的SEI可以通过阴离子的分解迅速修复。因此,可以保留颗粒状锂的沉积方式。然而,当产生较大的离子浓度梯度时,Li+-阴离子的相互作用减弱,配位阴离子的数量受到限制,难以维持SEI的持续修复。在这种情况下,阴离子衍生的SEI化学逐渐变成溶剂衍生的化学,这将锂沉积形态转变为针状树突状模式。
低温工作调节锂金属负极的电极液的合理设计
根据以上讨论,我们进一步探讨了同时针对低过电位和高锂离子可逆性的低温电解液的设计策略(图6a)。本质上,浓度过电位的降低需要较低的Li+/DME比值来促进锂盐的解离。相反,维持一个强大的Li+-阴离子相互作用对于有利的阴离子衍生的SEI形成需要一个足够高的Li+/DME比值。为了克服这种权衡困境,我们提出在电解质设计中分离快速的Li+转运的功能和持续的协调阴离子供应,在电解质设计中动态SEI形成。我们将DME/TTE(1/2byvol.)中LiFSI盐的浓度略微降低到0.8M,这促进了更高的离子电导率(0.31mScm-1),从而显著降低了电池的极化。此外,还引入了微量(0.05M)的强配位硝酸锂(LiNO3)来补偿Li+-阴离子聚集的损失。值得注意的是,增强的LiNO3(0.10M)将极大地影响电解质的低温离子电导率,甚至低于LHCE电解液。这与文献中观察到的传统含LiNO3的电解质的低温性能较差相一致。不同的是,本研究中开发的先进电解质在1.0mAcm-2下,Li|Cu电池的平均CE提高到98.40%,并降低了锂沉积过电位(图6b)。进一步采用滴定气相色谱(TGC)分析了循环电池的不可逆性来源(图6c)。结果表明,高级电解质中产生的死Li量仅为LHCE电解质的五分之一,这与它们明显的Li沉积密切相关。在LiNO3衍生的SEI化学条件下,球形的Li形态是一种典型的Li沉积模式。此外,结果表明,SEI-Li+容量损失相当的电解质,这可以解释为球形锂的表面积降低,LiNO3的高反应性可能会加重单位表面积的锂消耗。这两个因素的共同贡献最终导致了SEI-Li+容量的变化可以忽略不计。
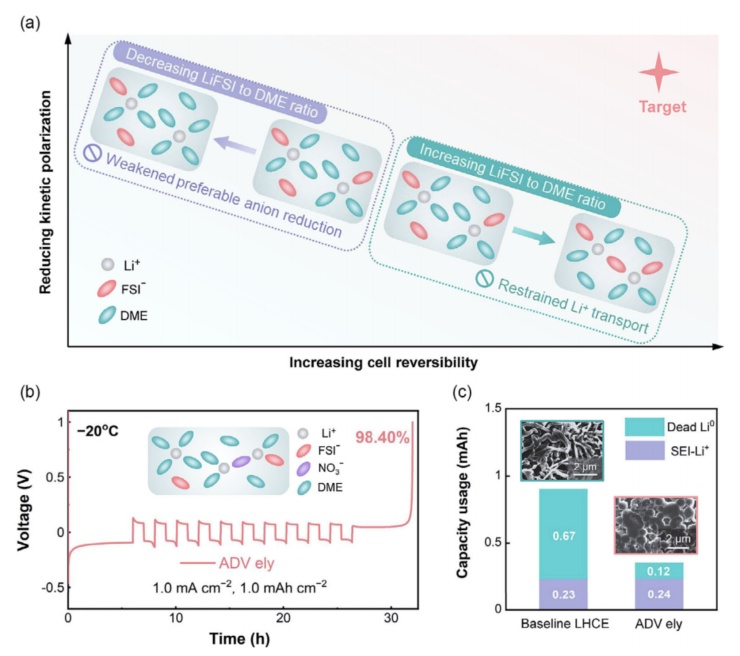
图6(a)低温电解质设计的示意图,同时针对低过电位和高锂可逆性,其中仅调节锂盐浓度是无法实现的。(b)利用先进的电解质测定Li|Cu半电池的平均CE。该插图说明了在优化电解质中形成的典型溶剂化结构。(c)在1.0mAcm-2下进行10个循环后的非活性Li组成的TGC结果,插图是LHCE和ADV电解质中各自的Li沉积形态。
Li|NCM523电采用有限的Li源(50μm)、高负载正极(≈3.3mAhcm-2)和有限的电解液(7.0μLmAh-1)用于低温循环(图7)。低温电池在初始循环中需要更长的激活期,这可能是由于电极润湿和SEI/CEI积累的动力学缓慢。在0.1C的低速率下,LHCE的电池可以循环60个周期,其容量保持率为80%(图7a)。相比之下,改善后的电解质在很大程度上增强了Li|NCM523电池的循环稳定性,在200个循环中可以观察到容量的衰减可以忽略不计(图7a)。进一步将电流速率提高到0.25C,两个电池的性能下降(图7b)。尽管如此,与对比样的短寿命相比(只有30个周期),使用优化的电解质的电池的使用寿命仍然延长了近3倍(图7b)。这种容量的衰减可以直接反映在电压曲线上(图7c,d),其中极化增强被确定为电池持续容量损失的根源。
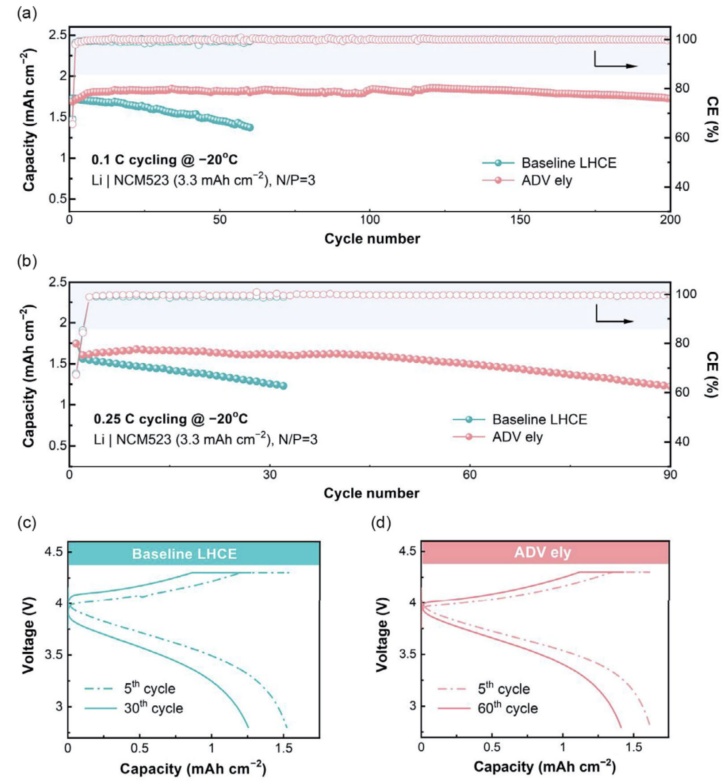
图7实际的Li|NCM523电池在(a)0.1C和(b)0.25C下循环的低温(-20°C)性能,以及使用c)LHCE和d)ADV电解质在0.25C下记录的电压曲线。
有趣的是,如果在-20°C下无法循环的电池重新进行室温循环,就可以消除如此大的过电位,容量损失几乎完全恢复。从这种非永久性容量损失现象可以推断,逐渐积累的非活性Li层内不断降解的电解质润湿应该是低温Li|NCM523电池极化增强的原因。这种由死锂和裂解的SEI组成的多孔层,一旦温度升高,就可以被电解质重新渗透,有助于减轻大极化和恢复电池容量。这说明低温电池的工作极化对非活性锂的产生更加敏感,这与电解质的物理性质(粘度、离子电导率等)的变化有关。
总结与展望
综上所述,本工作仔细研究了低温锂沉积的过电位属性,并以LHCE为模型平台,建立了过电位支配、SEI化学的动态演化和相应的锂利用效率之间的相关性。与常识不同的是,我们发现在零下工作温度下,浓度过电位主导了整个锂沉积过电位,这一方面限制了动力学性能,另一方面通过限制动态SEI形成过程中的阴离子分解,降低了锂沉积的均匀性。通过复杂地设计电解质化学,可以同时减少工作过电位和持续提供配位阴离子以形成动态SEI。因此,在-20°C下,Li|NCM523电池的CE沉积/溶解效率从95.93%明显提高,实际Li|NCM523电池提高到98.40%,寿命进一步延长了3倍以上。本工作揭示了低温工作锂金属负极极化和可逆性的实际因素,为实用锂金属电池在零下温度下稳定高效循环提供了新的设计原则。
(中国粉体网编辑整理/文正)
注:图片非商业用途,存在侵权告知删除!